AA Amyloidosis
AA amyloidosis occurs as a reaction to another illness, such as a chronic inflammatory disease or a chronic infection. Infections and inflammation cause the liver to produce a protein called SAA (serum amyloid A protein) at high levels. This is a normal reaction.
When inflammation goes on for a very long period of time, a small portion of the SAA protein, called AA protein, will separate from SAA and deposit in tissues as AA amyloid. Normally after an inflammatory reaction, the whole SAA protein is broken down to its amino acid components and recycled, as happens with all proteins. It is not known why in some individuals a partial breakdown of SAA to AA occurs.
These individuals usually have severe and chronic inflammatory conditions lasting several years. They develop AA amyloid deposits in all tissues, but the most common organ damage occurs in the kidneys. Some patients experience complications with their liver, spleen, thyroid, digestive tract, or heart. Any chronic inflammation that elevates the SAA protein for a long time has the potential to lead to AA amyloidosis.
Inflammatory diseases that are more commonly known to lead to AA amyloidosis are in the following categories. Rheumatologic diseases include rheumatoid arthritis, juvenile arthritis, ankylosing spondylitis, and psoriatic arthritis. Gastrointestinal inflammatory diseases, including Crohn’s disease and ulcerative colitis.
Chronic Infections, such as tuberculosis, bronchiectasis, osteomyelitis, or infections associated with cystic fibrosis, AIDS, and needle-using drug addiction that cause skin infections. Hematologic malignancies, including Hodgkin’s disease, renal cell carcinoma, and Castleman’s disease. Hereditary disorders that cause disruption of inflammatory genes, such as Familial Mediterranean Fever (FMF), TRAPS (tumour necrosis factor receptor-associated periodic syndrome), and others.
When inflammation goes on for a very long period of time, a small portion of the SAA protein, called AA protein, will separate from SAA and deposit in tissues as AA amyloid. Normally after an inflammatory reaction, the whole SAA protein is broken down to its amino acid components and recycled, as happens with all proteins. It is not known why in some individuals a partial breakdown of SAA to AA occurs.
These individuals usually have severe and chronic inflammatory conditions lasting several years. They develop AA amyloid deposits in all tissues, but the most common organ damage occurs in the kidneys. Some patients experience complications with their liver, spleen, thyroid, digestive tract, or heart. Any chronic inflammation that elevates the SAA protein for a long time has the potential to lead to AA amyloidosis.
Inflammatory diseases that are more commonly known to lead to AA amyloidosis are in the following categories. Rheumatologic diseases include rheumatoid arthritis, juvenile arthritis, ankylosing spondylitis, and psoriatic arthritis. Gastrointestinal inflammatory diseases, including Crohn’s disease and ulcerative colitis.
Chronic Infections, such as tuberculosis, bronchiectasis, osteomyelitis, or infections associated with cystic fibrosis, AIDS, and needle-using drug addiction that cause skin infections. Hematologic malignancies, including Hodgkin’s disease, renal cell carcinoma, and Castleman’s disease. Hereditary disorders that cause disruption of inflammatory genes, such as Familial Mediterranean Fever (FMF), TRAPS (tumour necrosis factor receptor-associated periodic syndrome), and others.
Symptoms
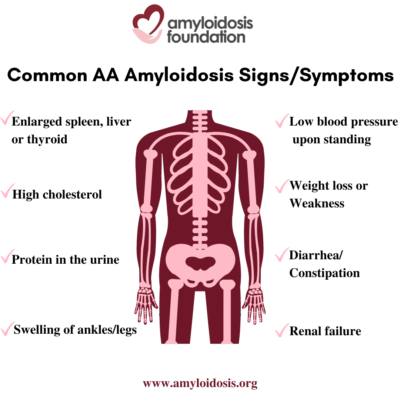
The term “nephrotic syndrome” refers to a group of symptoms that signal kidney problems. The first symptom of AA amyloidosis is usually protein in the urine. Often proteinuria (protein in the urine) becomes massive, and nephrotic syndrome develops. This means that patients are losing so much protein in their urine that they develop low levels of protein (albumin) in their blood, leading to swelling of the ankles and legs (edema). A high cholesterol level is also part of this syndrome. As a result, patients may develop renal failure and the need for dialysis.
AA amyloidosis involves other organs in addition to the kidneys. An enlarged spleen, enlarged liver, and enlarged thyroid are not uncommon. Autonomic neuropathy is frequent with symptoms of orthostatic hypotension (low blood pressure on standing), gastrointestinal atony (slowing of stomach emptying) and diarrhea or constipation. AA amyloid deposits in the heart causing congestive heart failure and arrhythmias (irregular heart beat) may develop later in the course of the disease.
Symptoms in a patient with AA amyloidosis can be misunderstood as symptoms that relate to their chronic infection or inflammation. At first, a patient may have symptoms such as weight loss, weakness, and swelling (edema). Since a patient’s primary disorder may also cause these problems, a doctor may assume that these are a result of their primary condition. This can delay further testing which would lead to a diagnosis of the secondary disease, AA amyloidosis.
In a reverse situation, AA amyloidosis may be found first, before another disease or condition is identified. For example, AA amyloidosis may be diagnosed as a result of nephrotic syndrome and then may lead to the investigation for an undiagnosed, underlying condition. So, although the AA amyloidosis was triggered by a primary disorder, it does not always mean that the primary disorder was previously discovered and diagnosed.
AA amyloidosis involves other organs in addition to the kidneys. An enlarged spleen, enlarged liver, and enlarged thyroid are not uncommon. Autonomic neuropathy is frequent with symptoms of orthostatic hypotension (low blood pressure on standing), gastrointestinal atony (slowing of stomach emptying) and diarrhea or constipation. AA amyloid deposits in the heart causing congestive heart failure and arrhythmias (irregular heart beat) may develop later in the course of the disease.
Symptoms in a patient with AA amyloidosis can be misunderstood as symptoms that relate to their chronic infection or inflammation. At first, a patient may have symptoms such as weight loss, weakness, and swelling (edema). Since a patient’s primary disorder may also cause these problems, a doctor may assume that these are a result of their primary condition. This can delay further testing which would lead to a diagnosis of the secondary disease, AA amyloidosis.
In a reverse situation, AA amyloidosis may be found first, before another disease or condition is identified. For example, AA amyloidosis may be diagnosed as a result of nephrotic syndrome and then may lead to the investigation for an undiagnosed, underlying condition. So, although the AA amyloidosis was triggered by a primary disorder, it does not always mean that the primary disorder was previously discovered and diagnosed.
Diagnosis
If a patient has previously been diagnosed with a chronic inflammatory disease or chronic infection and they develop high levels of protein in the urine or other associated AA symptoms, then the physician should test for AA amyloid deposition. When renal damage occurs, it can be clinically shown as proteinuria (protein found in the urine), nephrotic syndrome, or impairment of renal (kidney) function.
A test involving a 24-hour urine collection can be performed to look at the level of protein in the patient’s urine sample. Protein in the urine is an indication of kidney complications. If amyloidosis is suspected through this and other test results and associated symptoms, in most cases a biopsy of renal (kidney) tissue is recommended to get an accurate diagnosis.
This renal biopsy tissue is sent to a lab for Congo-red staining. The lab will stain the biopsy and, if it turns an apple green color under a ‘polarizing’ microscope, then amyloidosis is confirmed. Once this initial diagnosis has been determined, it is very important to find out the exact protein type in a positive tissue biopsy so that appropriate treatment can be recommended.
In order to identify the amyloid type, the most common diagnostic test is staining the tissue sample with antibodies that are specific for the major amyloid protein diseases, such as “anti-AA serum,” AL light chains, and anti-TTR. If it is the anti-AA serum result that is positive in this lab test, then AA amyloidosis is diagnosed. It is important to rule out other types because other amyloid diseases may involve the kidneys and those patients may also present with a high level of protein in their urine. However, another type of amyloid disease that is known for kidney involvement, such as AL amyloidosis, would require a different treatment regimen.
Once AA amyloidosis is confirmed it is important to look for the primary underlying inflammatory condition, if not already known. Then, the next step is to determine the extent of amyloid involvement in all organs and develop a plan for treatment. This is done by a careful physical examination, and laboratory studies of kidney and heart function.
A test involving a 24-hour urine collection can be performed to look at the level of protein in the patient’s urine sample. Protein in the urine is an indication of kidney complications. If amyloidosis is suspected through this and other test results and associated symptoms, in most cases a biopsy of renal (kidney) tissue is recommended to get an accurate diagnosis.
This renal biopsy tissue is sent to a lab for Congo-red staining. The lab will stain the biopsy and, if it turns an apple green color under a ‘polarizing’ microscope, then amyloidosis is confirmed. Once this initial diagnosis has been determined, it is very important to find out the exact protein type in a positive tissue biopsy so that appropriate treatment can be recommended.
In order to identify the amyloid type, the most common diagnostic test is staining the tissue sample with antibodies that are specific for the major amyloid protein diseases, such as “anti-AA serum,” AL light chains, and anti-TTR. If it is the anti-AA serum result that is positive in this lab test, then AA amyloidosis is diagnosed. It is important to rule out other types because other amyloid diseases may involve the kidneys and those patients may also present with a high level of protein in their urine. However, another type of amyloid disease that is known for kidney involvement, such as AL amyloidosis, would require a different treatment regimen.
Once AA amyloidosis is confirmed it is important to look for the primary underlying inflammatory condition, if not already known. Then, the next step is to determine the extent of amyloid involvement in all organs and develop a plan for treatment. This is done by a careful physical examination, and laboratory studies of kidney and heart function.
Treatment
Each amyloidosis type has a specific treatment. Early detection and timely treatment is a key factor. The type of treatment is based upon disease progression and seriousness of the patient’s organ, tissue and/or nerve involvement.
AA amyloidosis treatment plans include:
AA amyloidosis treatment plans include:
- Underlying disease treatment – continual management of the primary disease;
- Supportive treatment – treating patient symptoms and organ damage; and,
- Amyloid source treatment, when available – slowing down, or stopping, the overproduction of amyloid at the source of this secondary disease.
Underlying disease treatment
In AA amyloidosis, the most important therapy is to treat the underlying infection or inflammation in order to reduce the level of SAA protein, the precursor for the AA amyloid deposits. These treatments will vary depending on the underlying condition. The effectiveness of this treatment can be monitored by blood tests that measure inflammation in the blood, such as sedimentation rate and C-reactive protein levels. Some treatments that exist for inflammatory diseases could include surgery on the infection or tumor; drug therapies for rheumatoid arthritis; antibiotics for chronic infection; and the use of colchicine for FMF (Familial Mediterranean Fever), among others.
With effective treatment of the underlying inflammation amyloid deposits have been known to reduce and nephrotic syndrome can improve. However, if the kidney function has become significantly impaired, it rarely recovers.
With effective treatment of the underlying inflammation amyloid deposits have been known to reduce and nephrotic syndrome can improve. However, if the kidney function has become significantly impaired, it rarely recovers.
Supportive Treatment
Supportive treatment is very important for patients with AA amyloidosis. A team of specialists, including a nephrologist, cardiologist, and neurologist in addition to the primary physician is helpful to manage the disease development in the various organ systems.
Kidney damage is often a major health issue associated with AA amyloidosis. Regular blood and urine tests are recommended to monitor the patient’s renal (kidney) function. Managing proteinuria (protein in the urine) and nephrotic syndrome and keeping renal function from declining can delay the need for dialysis and improve the quality of life. Under the close supervision of the doctor, the supportive treatment that is often helpful for nephrotic syndrome includes an increase of protein in the diet, monitoring salt intake, and use of support hose. Other things that are very important when the kidney has amyloid deposits are maintaining normal blood pressure and avoiding dehydration.
It is important to note that the patient should not take any medication (prescription or over the counter) unless it has been approved by the nephrologist. When the kidney already has some damage, some medications can worsen this kidney damage. It can happen even when these same medications would be acceptable for normal kidneys. Therefore, it is very important that all patients with AA amyloidosis and renal involvement have a nephrologist on their medical team.
Dialysis is an option for patients with AA amyloidosis that are in renal failure, especially when other organ function is in good condition. A kidney transplant is an option in some cases, particularly if the associated inflammatory disease or chronic infection has been treated successfully.
Supportive treatment for autonomic neuropathy includes maintaining blood pressure when standing by using support hose, along with a slight increase of salt in the diet or medications that raise blood pressure.
Symptoms of gastric distress may be harder to manage, but frequent small meals and a diet lower in fat are often helpful. Medications can be used for diarrhea. An adjustment of the patient’s meal timing can also ease the onset of diarrhea.
Cardiac involvement needs to be watched closely by a cardiologist to manage treatment for congestive heart failure or arrhythmia.
Kidney damage is often a major health issue associated with AA amyloidosis. Regular blood and urine tests are recommended to monitor the patient’s renal (kidney) function. Managing proteinuria (protein in the urine) and nephrotic syndrome and keeping renal function from declining can delay the need for dialysis and improve the quality of life. Under the close supervision of the doctor, the supportive treatment that is often helpful for nephrotic syndrome includes an increase of protein in the diet, monitoring salt intake, and use of support hose. Other things that are very important when the kidney has amyloid deposits are maintaining normal blood pressure and avoiding dehydration.
It is important to note that the patient should not take any medication (prescription or over the counter) unless it has been approved by the nephrologist. When the kidney already has some damage, some medications can worsen this kidney damage. It can happen even when these same medications would be acceptable for normal kidneys. Therefore, it is very important that all patients with AA amyloidosis and renal involvement have a nephrologist on their medical team.
Dialysis is an option for patients with AA amyloidosis that are in renal failure, especially when other organ function is in good condition. A kidney transplant is an option in some cases, particularly if the associated inflammatory disease or chronic infection has been treated successfully.
Supportive treatment for autonomic neuropathy includes maintaining blood pressure when standing by using support hose, along with a slight increase of salt in the diet or medications that raise blood pressure.
Symptoms of gastric distress may be harder to manage, but frequent small meals and a diet lower in fat are often helpful. Medications can be used for diarrhea. An adjustment of the patient’s meal timing can also ease the onset of diarrhea.
Cardiac involvement needs to be watched closely by a cardiologist to manage treatment for congestive heart failure or arrhythmia.