AL Amyloidosis
In the United States, AL amyloidosis is the most common type, with approximately 4,500 new cases diagnosed every year. It usually affects people from ages 50-80, although there are a few cases of people being diagnosed as early as their late 20s. About two-thirds of the patients are male.
AL amyloidosis is caused by a bone marrow disorder. The bone marrow in the center of bones produces cells in the blood system, including “plasma cells.” These plasma cells are the part of the immune system that makes antibodies for fighting infections. The term “immunoglobulin” refers to the class of proteins that function as antibodies. Immunoglobulins are composed of four protein chains: two light chains, either kappa or lambda light chains, and two heavy chains, of which there are several types.
These proteins are produced by the plasma cells in the bone marrow. In AL patients, these plasma cells produce an abnormal antibody (immunoglobulin) protein. For AL amyloidosis, it is the “light chains” that become misfolded, and the abnormal, misfolded result is the forming of amyloid. With AL amyloidosis, the “A” is for amyloid and the “L” is for light chain.
These misfolded amyloid proteins are deposited in and around tissues, nerves and organs. As the amyloid builds up in an organ, nerve or tissue, it gradually causes damage and affects their function. Each amyloidosis patient has a different pattern of amyloid deposition in their body. It often affects more than one organ. AL amyloidosis does not affect the brain.
AL amyloidosis is caused by a bone marrow disorder. The bone marrow in the center of bones produces cells in the blood system, including “plasma cells.” These plasma cells are the part of the immune system that makes antibodies for fighting infections. The term “immunoglobulin” refers to the class of proteins that function as antibodies. Immunoglobulins are composed of four protein chains: two light chains, either kappa or lambda light chains, and two heavy chains, of which there are several types.
These proteins are produced by the plasma cells in the bone marrow. In AL patients, these plasma cells produce an abnormal antibody (immunoglobulin) protein. For AL amyloidosis, it is the “light chains” that become misfolded, and the abnormal, misfolded result is the forming of amyloid. With AL amyloidosis, the “A” is for amyloid and the “L” is for light chain.
These misfolded amyloid proteins are deposited in and around tissues, nerves and organs. As the amyloid builds up in an organ, nerve or tissue, it gradually causes damage and affects their function. Each amyloidosis patient has a different pattern of amyloid deposition in their body. It often affects more than one organ. AL amyloidosis does not affect the brain.
Symptoms
The symptoms of AL amyloidosis vary by patient. Initially, the symptoms can be minor or similar to those of many other conditions or systemic diseases. Some symptoms can announce themselves quickly and be very noticeable. For each patient, the symptoms will depend on which organs are affected by the amyloid deposits. It also depends on the degree that the organ function is impaired. Fatigue, weight loss and swelling are the most common symptoms.
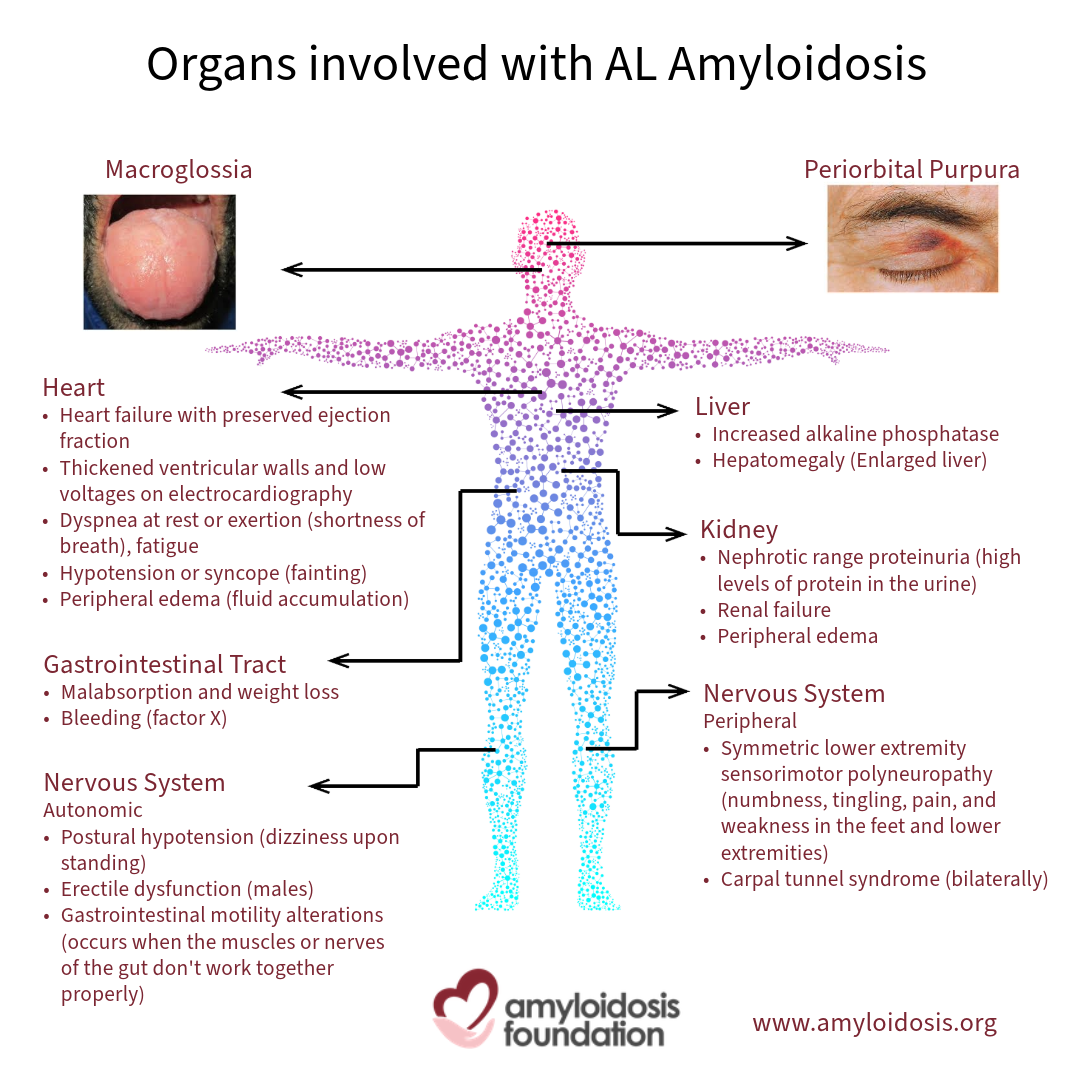
Impairment of many organs, nerves and soft tissues can cause symptoms, among them the kidneys, heart, the GI tract (the digestive system) and the nervous system.
Impairment of many organs, nerves and soft tissues can cause symptoms, among them the kidneys, heart, the GI tract (the digestive system) and the nervous system.
The Kidneys
Chronic kidney disease is common in patients with AL amyloidosis. Amyloid deposits in the kidneys can affect how they filter toxins and proteins in the blood. This may result in a condition called nephrotic syndrome, where there is excess protein in the urine and the lower legs can become swollen (also called “edema”). Swelling can affect the belly, arms, and lungs as well. In some cases, the amyloid deposits will cause the kidneys to lose the ability to purify the blood, which can lead to kidney failure; also known as “renal” failure. These patients may need dialysis to replace the function of the kidneys.
The Heart
Amyloid deposits in the heart can cause it to become unusually thickened and stiff, making it unable to function efficiently. This results in shortness of breath, which may occur with only minor activity. Amyloid can also affect the electrical system of the heart, causing the normal heartbeat to speed up or slow down. This is known as arrhythmia.
The Digestive System
The digestive system is also called the gastrointestinal tract (or GI tract). Amyloid deposits in the digestive system can cause nausea, diarrhea or constipation, weight loss, loss of appetite, or a feeling of fullness in the stomach after eating small amounts.
The Nervous System
Amyloid deposits can affect the nerves of the hands, feet and lower legs and may cause pain, numbness and tingling. A loss of sensitivity to temperature may also occur. This is called peripheral neuropathy. Nerves that control blood pressure, heart rate, bowel motility, erectile function, and other body functions can also be affected, causing a variety of symptoms including dizziness when standing too quickly, nausea and diarrhea. This is called autonomic neuropathy.
Other Symptoms
There are other symptoms that are common and may have been present for some time before diagnosis, such as chronic fatigue and weakness. Some patients with AL amyloidosis experience purpura, which is bruising around the eyes or other skin areas.
Swelling may develop and cause symptoms as a result of the amyloid deposits. For example, patients may have carpal tunnel syndrome, where amyloid deposits in the wrist area can squeeze and irritate the nerve, causing tingling and numbness in the fingers and thumb. Deposits in the tongue may lead to a swollen and enlarged tongue, called “macroglossia.” In addition, some patients experience the “shoulder pad” symptom; where the patient notices an enlargement of the shoulders, causing restriction in the joint due to swelling and/or amyloid deposits in the surrounding tissues.
One significant, but less common, symptom is that some patients can develop bleeding or clotting problems.
Swelling may develop and cause symptoms as a result of the amyloid deposits. For example, patients may have carpal tunnel syndrome, where amyloid deposits in the wrist area can squeeze and irritate the nerve, causing tingling and numbness in the fingers and thumb. Deposits in the tongue may lead to a swollen and enlarged tongue, called “macroglossia.” In addition, some patients experience the “shoulder pad” symptom; where the patient notices an enlargement of the shoulders, causing restriction in the joint due to swelling and/or amyloid deposits in the surrounding tissues.
One significant, but less common, symptom is that some patients can develop bleeding or clotting problems.
Diagnosis
Diagnostic testing for AL amyloidosis involves blood tests, urine tests and biopsies. Blood and/or urine tests can indicate signs of the amyloid protein, but only bone marrow tests or other small biopsy samples of tissue or organs can positively confirm the diagnosis of amyloidosis. Some tests are only done once to establish a diagnosis of AL amyloidosis, while others will be repeated to monitor disease progression and response to therapy.
Blood and Urine Tests
Blood and urine tests should be performed to help verify the diagnosis. They can also aid in discovering which organs are involved and how much they are compromised. These tests may include:
A 24-hour urine collection to look at the level of protein in your urine sample. Excess protein in the urine may be an indication of kidney involvement. The level of ALP (an enzyme called “alkaline phosphatase”) in your regular blood workup. Blood tests to look for stress and strain on the heart are useful in many forms of heart disease, including AL amyloidosis. The cardiac biomarkers that are used include troponin T or troponin I, and NT-proBNP (which stands for N-terminal pro-brain natriuretic peptide) or BNP (brain natriuretic peptide). Different laboratories use one versus the other. Tests for abnormal antibody (immunoglobulin) proteins in the blood include the Freelite Assay, which shows the level of kappa and lambda light chains in a separate blood test. The Freelite Assay test is often referred to as FLC, which is an abbreviation for free light chains. Another test for abnormal immunoglobulin can be done with blood and/or urine. It is called “immunofixation electrophoresis.”
These blood and urine tests can help with the diagnosis and used often while monitoring response to treatment.
A 24-hour urine collection to look at the level of protein in your urine sample. Excess protein in the urine may be an indication of kidney involvement. The level of ALP (an enzyme called “alkaline phosphatase”) in your regular blood workup. Blood tests to look for stress and strain on the heart are useful in many forms of heart disease, including AL amyloidosis. The cardiac biomarkers that are used include troponin T or troponin I, and NT-proBNP (which stands for N-terminal pro-brain natriuretic peptide) or BNP (brain natriuretic peptide). Different laboratories use one versus the other. Tests for abnormal antibody (immunoglobulin) proteins in the blood include the Freelite Assay, which shows the level of kappa and lambda light chains in a separate blood test. The Freelite Assay test is often referred to as FLC, which is an abbreviation for free light chains. Another test for abnormal immunoglobulin can be done with blood and/or urine. It is called “immunofixation electrophoresis.”
These blood and urine tests can help with the diagnosis and used often while monitoring response to treatment.
Echocardiogram and Imaging
The echocardiogram (also called “echo”) is an ultrasound of the heart. A doctor can look for amyloid deposits in the heart, while viewing the size and shape of it and the location and extent of any impact of amyloid.
Recently, other imaging tests for the heart have also shown to be useful. One test is the MRI (magnetic resonance imaging), and, in this instance, is also referred to as CMR (for cardiac magnetic resonance). Pyrophosphate scanning, a nuclear medicine test, is also used to evaluate whether an unusual type of abnormality of heart muscle function (“cardiomyopathy”) is present. Current data suggests this scan may be useful in distinguishing different types of amyloid heart disease.
Recently, other imaging tests for the heart have also shown to be useful. One test is the MRI (magnetic resonance imaging), and, in this instance, is also referred to as CMR (for cardiac magnetic resonance). Pyrophosphate scanning, a nuclear medicine test, is also used to evaluate whether an unusual type of abnormality of heart muscle function (“cardiomyopathy”) is present. Current data suggests this scan may be useful in distinguishing different types of amyloid heart disease.
Tissue Biopsy
A tissue biopsy involves the removal of a small sample of tissue to find evidence of amyloid deposits. Any kind of tissue or organ biopsy must be sent to a lab for microscopic examination, where the tissue is stained with a dye called “Congo-red stain.” After putting it under a microscope, amyloid protein is discovered if it turns an apple-green color, resulting in a diagnosis of amyloidosis. Possible areas for less invasive biopsies include:
A fat pad biopsy (from under the skin in the abdomen); Labial salivary gland biopsy (the inner lip); and, Skin or bone marrow.
With combinations of these blood and urine tests and tissue biopsies, a positive diagnosis can be confirmed in a high percentage of patients.
A fat pad biopsy (from under the skin in the abdomen); Labial salivary gland biopsy (the inner lip); and, Skin or bone marrow.
With combinations of these blood and urine tests and tissue biopsies, a positive diagnosis can be confirmed in a high percentage of patients.
Bone Marrow Aspirate and Biopsy
There are two types of bone marrow tests that may be performed. These involve the removal of some liquid bone marrow (a bone marrow aspirate) and/or the removal of a 1 – 2 cm core of bone marrow tissue in one piece (a bone marrow biopsy). These samples can help to determine the percentage of amyloid producing plasma cells, and when tested in the lab they can assist in identifying whether the abnormal plasma cells are producing kappa or lambda light chains.
Organ Biopsy
If amyloidosis is still suspected but biopsies of the bone marrow, fat pad, lip or skin sites turn up negative, then a surgical biopsy of the organ that is indicating symptoms should be performed and sent to a lab. Biopsy samples may be taken from the:
Liver Kidney Nerve Heart Gut (stomach or intestines).
If any biopsy result shows a positive diagnosis for amyloidosis, then it is essential to also determine the accurate type of amyloid protein that is involved. In this case, the type of AL amyloidosis must be confirmed, showing a bone marrow disorder with light chain involvement, also known as a “plasma cell dyscrasia.”
Liver Kidney Nerve Heart Gut (stomach or intestines).
If any biopsy result shows a positive diagnosis for amyloidosis, then it is essential to also determine the accurate type of amyloid protein that is involved. In this case, the type of AL amyloidosis must be confirmed, showing a bone marrow disorder with light chain involvement, also known as a “plasma cell dyscrasia.”
Treatment
Treatment for AL amyloidosis is tailored to the patient with their individual health in mind. The type of treatment is based upon disease progression and seriousness of the patient’s organ, tissue, and nerve involvement.
Today’s treatment plans are two-fold:
Supportive treatment – treating your symptoms and organ damage; and, Source treatment – slowing down, or stopping, the overproduction of amyloid at the source of the disease.
The doctor will need to prescribe the supportive treatment that the patient needs as well as source treatment for the disease itself.
Today’s treatment plans are two-fold:
Supportive treatment – treating your symptoms and organ damage; and, Source treatment – slowing down, or stopping, the overproduction of amyloid at the source of the disease.
The doctor will need to prescribe the supportive treatment that the patient needs as well as source treatment for the disease itself.
Supportive Treatment
Supportive treatment is helpful for various symptoms, including cardiac and kidney problems, and can change the quality of life for many people.
For example, gastrointestinal dysfunction may require treatment for symptoms that include poor nutritional health, diarrhea or constipation, and nausea or vomiting. Doctors can prescribe medications to help with these symptoms to lessen the pain and the symptom itself.
Management of heart problems, heart failure, and kidney dialysis can make a significant improvement in a patient’s quality of life. Treatment for heart and kidney failure in amyloidosis is different from organ problems due to other diseases. It is important for a patient to consult with specialists that are aware of what medications are, and are not, safe for amyloidosis patients.
Reversing any damage to the organs and other parts of the body is difficult to achieve. If treatment begins during the early onset of clinical symptoms, the overall success rate is higher, so early detection is essential.
For example, gastrointestinal dysfunction may require treatment for symptoms that include poor nutritional health, diarrhea or constipation, and nausea or vomiting. Doctors can prescribe medications to help with these symptoms to lessen the pain and the symptom itself.
Management of heart problems, heart failure, and kidney dialysis can make a significant improvement in a patient’s quality of life. Treatment for heart and kidney failure in amyloidosis is different from organ problems due to other diseases. It is important for a patient to consult with specialists that are aware of what medications are, and are not, safe for amyloidosis patients.
Reversing any damage to the organs and other parts of the body is difficult to achieve. If treatment begins during the early onset of clinical symptoms, the overall success rate is higher, so early detection is essential.
Source Treatment
The U.S. Food and Drug Administration (FDA) has approved the use of DARZALEX FASPRO® (daratumumab and hyaluronidase-fihj), a subcutaneous formulation of daratumumab, in combination with bortezomib, cyclophosphamide, and dexamethasone (D-VCd) for the treatment of adult patients with newly diagnosed light chain (AL) amyloidosis.
Stem Cell Transplant
In the United States, a stem cell transplant (SCT) is often the preferred therapy, as it can provide long-term control of the underlying disease. However, only a minority of AL patients are eligible for this. For the majority of AL amyloidosis patients, and in many other countries outside the U.S., other chemotherapy-based treatments are considered. These treatment regimens are tailored for each patient and based upon the patients’ organ function, symptoms, and preferences.
Combination Therapy
Patients with AL amyloidosis have benefited from the recent development of new drugs for myeloma, many of which work effectively on the plasma cells that cause AL amyloidosis. Many drug combinations are more effective than single drugs in attacking the abnormal plasma cells and the dosage is tailored to each individual patient, to enable the best course of treatment and possible outcome. Categories of drugs that may be useful include:
Traditional Chemotherapy Drugs:
- Melphalan, Cyclophosphamide (Cytoxan™), Bendamustine (Treanda™)
- Drugs called “proteasome inhibitors”: Bortezomib (Velcade™), MLN9708 (Ixazomib™), Carfilzomib (Kyprolis™)
- Drugs called “immunomodulators”: Thalidomide (Thalidomid™), Lenalidomide (Revlimid™), Pomalidomide (Pomalyst™)
Note that some of these drugs are still considered investigational for AL amyloidosis, and should not be used outside of a sponsored clinical trial. Also, most of them are given with a steroid such as dexamethasone, which seems to increase their effectiveness.
FAQ
Why is it called AL amyloidosis?
AL amyloidosis used to be called “Primary” amyloidosis. This is no longer an accepted name for this form of amyloidosis, which is caused by light chains from the bone marrow.
Connecting the dots with your body’s biology can be difficult to follow. Your bone marrow makes platelets and red and white blood cells. Plasma cells are the type of white blood cell that makes antibodies (which are also proteins).
These antibodies, or proteins, can also be called immunoglobulin. If you have been diagnosed with AL amyloidosis, you will often hear your doctor refer to immunoglobulin. Immunoglobulin has a basic structure of heavy chains and two small light chains. Light chains that get “free” from an antibody and misfold are the culprits for disease in AL amyloidosis, and during the “free” stage can be measured in the FreeLite assay test.
With AL amyloidosis, the “A” is for amyloid and the “L” is for light chain.
Connecting the dots with your body’s biology can be difficult to follow. Your bone marrow makes platelets and red and white blood cells. Plasma cells are the type of white blood cell that makes antibodies (which are also proteins).
These antibodies, or proteins, can also be called immunoglobulin. If you have been diagnosed with AL amyloidosis, you will often hear your doctor refer to immunoglobulin. Immunoglobulin has a basic structure of heavy chains and two small light chains. Light chains that get “free” from an antibody and misfold are the culprits for disease in AL amyloidosis, and during the “free” stage can be measured in the FreeLite assay test.
With AL amyloidosis, the “A” is for amyloid and the “L” is for light chain.
Is there more than one type of AL amyloidosis?
If you have been diagnosed with AL amyloidosis, there are two kinds of “L” (light chains) that you should know. The light chains are represented by two Greek letters: kappa (κ) and lambda (λ). So, a patient with AL amyloidosis may have a disease emphasis with either the kappa or lambda type of light chain. Lambda (λ) is more common than kappa (κ). However, at this time, the type of light chain identified with a patient’s AL amyloidosis does not change the diagnosis or treatment.
What is the underlying cause of AL amyloidosis?
While the abnormal light chain is responsible for the deposits and organ damage in the disease, it is believed that the plasma cell they come from is the real underlying cause of disease. Treatments currently target the plasma cells, but in addition, investigational treatments to target the light chain fibrils are also being developed. These might work together very well.
AL amyloidosis is not contagious, is not secondary to other diseases, and is not considered to be hereditary. However, there are very rare families that have an increased incidence of blood diseases including myeloma, lymphoma, “MGUS” (monoclonal gammopathy of undetermined significance), and AL amyloidosis. Note that in Alzheimer’s disease, there are “amyloid plaques” in the brain, but these are not formed from light chains, and AL patients have no increased susceptibility to Alzheimer’s disease, compared with the general population.
From the current available data, it has been determined that approximately 2/3 of the AL amyloidosis patients are male. The age range of AL amyloidosis usually affects people from ages 50-80, although there are cases of people being diagnosed and treated at younger ages, with less than 5% of the patients being under 40 years old.
AL amyloidosis is not contagious, is not secondary to other diseases, and is not considered to be hereditary. However, there are very rare families that have an increased incidence of blood diseases including myeloma, lymphoma, “MGUS” (monoclonal gammopathy of undetermined significance), and AL amyloidosis. Note that in Alzheimer’s disease, there are “amyloid plaques” in the brain, but these are not formed from light chains, and AL patients have no increased susceptibility to Alzheimer’s disease, compared with the general population.
From the current available data, it has been determined that approximately 2/3 of the AL amyloidosis patients are male. The age range of AL amyloidosis usually affects people from ages 50-80, although there are cases of people being diagnosed and treated at younger ages, with less than 5% of the patients being under 40 years old.
Why is AL amyloidosis sometimes linked with Multiple Myeloma?
AL may occasionally be associated with myeloma, because like AL amyloidosis, multiple myeloma affects the plasma cells inside the bone marrow.
In myeloma, there are so many plasma cells, that it clearly is a bone marrow cancer. It does not always form lesions, lumps or tumors, but the myeloma cells break down and expand inside the bone marrow, which can cause bone pain and fractures. This is not the case for AL amyloidosis. Although AL is a related disease, it is not clearly classified as a bone marrow cancer like myeloma or leukemia. There are myeloma and lymphoma patients who do develop AL amyloidosis.
Approximately 15% of those diagnosed with multiple myeloma will also acquire AL amyloidosis. Even if a patient has myeloma associated with their amyloidosis, the treatments for AL amyloidosis are similar to those for myeloma.
In myeloma, there are so many plasma cells, that it clearly is a bone marrow cancer. It does not always form lesions, lumps or tumors, but the myeloma cells break down and expand inside the bone marrow, which can cause bone pain and fractures. This is not the case for AL amyloidosis. Although AL is a related disease, it is not clearly classified as a bone marrow cancer like myeloma or leukemia. There are myeloma and lymphoma patients who do develop AL amyloidosis.
Approximately 15% of those diagnosed with multiple myeloma will also acquire AL amyloidosis. Even if a patient has myeloma associated with their amyloidosis, the treatments for AL amyloidosis are similar to those for myeloma.
What damage can AL amyloidosis do to your body?
When these light chain proteins assemble incorrectly, they are called abnormal, or misfolded, protein. When they travel in the body, they become rigid and insoluble (unable to dissolve), and are made up of tiny fibers (fibrils). This causes these misfolded proteins to gang up and cluster together, leaving deposits in and around body organs, connective tissues, muscles, and nerves. This interrupts the body’s normal function and causes symptoms. If this cluster of deposits is not slowed or stopped, then organ failure is possible.
The pattern of where the amyloid deposits in the body are often different when comparing one patient to another because every person is unique. Any number of organs and/or parts of the body can be affected, with the severity of damage varying from organ to organ as well as person-to-person.
In AL amyloidosis, common combinations of organ involvement include: heart/kidney; heart/GI tract; and, kidney/peripheral nerves — but almost any combination is possible. It is important to note that AL amyloidosis does not affect the brain.
The pattern of where the amyloid deposits in the body are often different when comparing one patient to another because every person is unique. Any number of organs and/or parts of the body can be affected, with the severity of damage varying from organ to organ as well as person-to-person.
In AL amyloidosis, common combinations of organ involvement include: heart/kidney; heart/GI tract; and, kidney/peripheral nerves — but almost any combination is possible. It is important to note that AL amyloidosis does not affect the brain.
How is AL amyloidosis diagnosed and what tests are involved?
Some symptoms can represent much milder ailments, even something like the flu, for example, gastric distress or diarrhea. This is why you and your internist or family doctor must be alert for continuing, or increasing, symptoms. If your symptoms don’t stop, then keep asking questions of your medical team so they can give you an accurate diagnosis.
Blood and/or urine tests can indicate signs of the amyloid protein, but only bone marrow tests or other small biopsy samples of tissue or organs can positively confirm the diagnosis of amyloidosis.
Blood and/or urine tests can indicate signs of the amyloid protein, but only bone marrow tests or other small biopsy samples of tissue or organs can positively confirm the diagnosis of amyloidosis.
Blood and Urine Tests
Getting a quick and timely diagnosis is vital. For example, the kidney is involved in approximately 65% of AL amyloidosis cases, so if a high concentration of protein is seen in your urine on a lab test, it is a signal to dig deeper when looking for a diagnosis.
Some tests can be used for diagnosis as well as for monitoring a patient’s response to treatment, although organ responses are typically much slower than hematologic (blood) responses. These tests include:
A 24-hour urine collection to look at the level of protein in your urine sample. Excess protein in the urine may be an indication of kidney involvement. Urine immunofixation is a lab test to look for clonal immunoglobulin proteins in urine. You may hear the term “Bence Jones proteins.” These are free immunoglobulin light chains that are found in high levels in the urine.
1. Testing for the level of ALP (an enzyme called “alkaline phosphatase”) should be included in your regular blood lab workup. An ALP test measures the amount of this enzyme in the blood. ALP is made mostly in the liver, but is also produced in bone and other parts of the body and can signal liver or bone problems. Certain conditions cause large amounts of ALP in the blood, so if your ALP level is high, it follows that more tests should be done to find the cause.
2. Tests for abnormal antibody (immunoglobulin) proteins in the blood include the Freelite® (Serum Free Light Chain) Assay Test (also known as FLC test). A blood lab measurement of the light chains can be very helpful in initial diagnostic testing and continuous monitoring of the disease. This blood test is relatively inexpensive and it measures both the kappa (κ) and lambda (λ) light chain production as well as the ratio between them.
3. If heart involvement is suspected, then blood tests for heart biomarkers can aid in determining if a patient has signs of heart tissue strain or damage in their blood. The results of these tests can be used as “markers” (or “biomarkers”) to first determine the extent of any damage, and then can be used regularly to monitor any future problems. Two troponin tests can be done — cardiac serum troponin T and serum troponin I. The other important biomarker is N-terminal pro-brain natriuretic peptide (NT-proBNP) or brain natriuretic peptide (BNP).
4. Different laboratories use one versus another.
Cardiac troponins T (cTnT) and I (cTnI) are released when the heart muscle has had some injury. In general, the more damage there is to the heart, the greater the amount of troponin T and I there will be in the blood. NT-proBNP is another “biomarker” that should be performed, especially if a person has symptoms such as swelling in the legs (edema), difficulty breathing, shortness of breath, and fatigue. It is used to detect heart stress or strain. This blood test can be useful in distinguishing fluid in the lungs due to congestive heart failure (in which it would be elevated) or from lung or pleural disease (in which case it should be normal or near normal). The pleurae are the linings of the lung, and can be involved with amyloid.
These biomarker blood tests can be affected by changes in kidney function, drugs, and other causes. They should be interpreted in the context of other tests of cardiac function, such as an echocardiogram or cardiac magnetic resonance imaging.
5. Another lab test for abnormal immunoglobulin proteins can be done with blood and/or urine. It is called “immunofixation electrophoresis.” This test helps to identify if one certain immunoglobulin is being over produced. Electrophoresis is a lab test where an electric current is applied to a substance (in this case, blood or urine) to separate it into its component parts. With immunofixation electrophoresis, it is the immunoglobulins that are being separated, then they are stained for further diagnostic results.
The above blood or urine test results can be informative clues if they show something that is not within normal range. They are the beginning phase of diagnosing AL (light chain) amyloidosis, but do not provide 100% accuracy for diagnosing a light chain abnormality. More tests will be necessary if any type of amyloidosis is suspected.
Some tests can be used for diagnosis as well as for monitoring a patient’s response to treatment, although organ responses are typically much slower than hematologic (blood) responses. These tests include:
A 24-hour urine collection to look at the level of protein in your urine sample. Excess protein in the urine may be an indication of kidney involvement. Urine immunofixation is a lab test to look for clonal immunoglobulin proteins in urine. You may hear the term “Bence Jones proteins.” These are free immunoglobulin light chains that are found in high levels in the urine.
1. Testing for the level of ALP (an enzyme called “alkaline phosphatase”) should be included in your regular blood lab workup. An ALP test measures the amount of this enzyme in the blood. ALP is made mostly in the liver, but is also produced in bone and other parts of the body and can signal liver or bone problems. Certain conditions cause large amounts of ALP in the blood, so if your ALP level is high, it follows that more tests should be done to find the cause.
2. Tests for abnormal antibody (immunoglobulin) proteins in the blood include the Freelite® (Serum Free Light Chain) Assay Test (also known as FLC test). A blood lab measurement of the light chains can be very helpful in initial diagnostic testing and continuous monitoring of the disease. This blood test is relatively inexpensive and it measures both the kappa (κ) and lambda (λ) light chain production as well as the ratio between them.
3. If heart involvement is suspected, then blood tests for heart biomarkers can aid in determining if a patient has signs of heart tissue strain or damage in their blood. The results of these tests can be used as “markers” (or “biomarkers”) to first determine the extent of any damage, and then can be used regularly to monitor any future problems. Two troponin tests can be done — cardiac serum troponin T and serum troponin I. The other important biomarker is N-terminal pro-brain natriuretic peptide (NT-proBNP) or brain natriuretic peptide (BNP).
4. Different laboratories use one versus another.
Cardiac troponins T (cTnT) and I (cTnI) are released when the heart muscle has had some injury. In general, the more damage there is to the heart, the greater the amount of troponin T and I there will be in the blood. NT-proBNP is another “biomarker” that should be performed, especially if a person has symptoms such as swelling in the legs (edema), difficulty breathing, shortness of breath, and fatigue. It is used to detect heart stress or strain. This blood test can be useful in distinguishing fluid in the lungs due to congestive heart failure (in which it would be elevated) or from lung or pleural disease (in which case it should be normal or near normal). The pleurae are the linings of the lung, and can be involved with amyloid.
These biomarker blood tests can be affected by changes in kidney function, drugs, and other causes. They should be interpreted in the context of other tests of cardiac function, such as an echocardiogram or cardiac magnetic resonance imaging.
5. Another lab test for abnormal immunoglobulin proteins can be done with blood and/or urine. It is called “immunofixation electrophoresis.” This test helps to identify if one certain immunoglobulin is being over produced. Electrophoresis is a lab test where an electric current is applied to a substance (in this case, blood or urine) to separate it into its component parts. With immunofixation electrophoresis, it is the immunoglobulins that are being separated, then they are stained for further diagnostic results.
The above blood or urine test results can be informative clues if they show something that is not within normal range. They are the beginning phase of diagnosing AL (light chain) amyloidosis, but do not provide 100% accuracy for diagnosing a light chain abnormality. More tests will be necessary if any type of amyloidosis is suspected.
Four Biopsies
Nonsurgical biopsies are also called minimally invasive biopsies. They can be performed individually or in combination. These can be done by taking small samples of the fat pad in the stomach, the inner lip area, the skin or inside the bone marrow.
1. With a “fat pad biopsy,” your doctor will clean your skin around your stomach and, with a needle, take a small piece of the ‘fat pad’ under the skin. What is taken from the needle is sent to a lab for analysis. Approximately 80% of the time you will get a clear diagnosis with a fat pad biopsy, however, this is still not at 100%. If it comes up negative, the doctor will want to continue with other diagnostic procedures to be certain of the diagnosis.
2. Another biopsy site is the ‘labial salivary gland’ where a small tissue sample is taken from an area in the inner lip.
3. Testing of the skin involves a biopsy that is sent to a lab for analysis.
4. A bone marrow biopsy takes a small amount of bone as well as the marrow (cells and fluid) from inside the bone. These samples can help to determine the percentage of plasma cells, and when tested in the lab they can aid in identifying whether the abnormal plasma cells are producing kappa or lambda light chains.
Interestingly, if you get a negative result from a fat pad biopsy, the doctor may want to consider performing an additional labial salivary gland biopsy. This is because it was noted in a medical article that 60% of the people who got an additional labial salivary gland biopsy (after they tested negative with the fat pad biopsy) came up positive for amyloid. So, if these two tests are performed back-to-back, the range of accuracy rises to about 91%. That means that the more invasive surgery and biopsy of an inner organ can be reduced to approximately 9% of patients.
1. With a “fat pad biopsy,” your doctor will clean your skin around your stomach and, with a needle, take a small piece of the ‘fat pad’ under the skin. What is taken from the needle is sent to a lab for analysis. Approximately 80% of the time you will get a clear diagnosis with a fat pad biopsy, however, this is still not at 100%. If it comes up negative, the doctor will want to continue with other diagnostic procedures to be certain of the diagnosis.
2. Another biopsy site is the ‘labial salivary gland’ where a small tissue sample is taken from an area in the inner lip.
3. Testing of the skin involves a biopsy that is sent to a lab for analysis.
4. A bone marrow biopsy takes a small amount of bone as well as the marrow (cells and fluid) from inside the bone. These samples can help to determine the percentage of plasma cells, and when tested in the lab they can aid in identifying whether the abnormal plasma cells are producing kappa or lambda light chains.
Interestingly, if you get a negative result from a fat pad biopsy, the doctor may want to consider performing an additional labial salivary gland biopsy. This is because it was noted in a medical article that 60% of the people who got an additional labial salivary gland biopsy (after they tested negative with the fat pad biopsy) came up positive for amyloid. So, if these two tests are performed back-to-back, the range of accuracy rises to about 91%. That means that the more invasive surgery and biopsy of an inner organ can be reduced to approximately 9% of patients.
Organ Biopsy
A small sample of an organ that is showing symptoms should be biopsied if amyloidosis is still suspected. This is often done after biopsies of the fat pad, lip, skin, or bone marrow (as mentioned above) turn up negative.
For example, if both fat pad and labial salivary gland biopsies are negative, but there is a high level of protein found in the patient’s urine sample, then a biopsy of a kidney is the next step.
In all biopsy cases, both nonsurgical and surgical, the lab analysis is the same. If an amyloid fibril has been deposited into body tissue, it can be discovered in a biopsy when the biopsy is chemically tested and viewed through a microscope with polarized light. A stain called the “Congo-red stain” is put on the biopsy tissue and if the examiner then sees the light wave change to an apple-green color (called “birefringence”) then amyloid is confirmed.
If any biopsy specimen results in a positive diagnosis for amyloidosis, then an accurate typing of the kind of amyloid protein is the next important step.
For example, if both fat pad and labial salivary gland biopsies are negative, but there is a high level of protein found in the patient’s urine sample, then a biopsy of a kidney is the next step.
In all biopsy cases, both nonsurgical and surgical, the lab analysis is the same. If an amyloid fibril has been deposited into body tissue, it can be discovered in a biopsy when the biopsy is chemically tested and viewed through a microscope with polarized light. A stain called the “Congo-red stain” is put on the biopsy tissue and if the examiner then sees the light wave change to an apple-green color (called “birefringence”) then amyloid is confirmed.
If any biopsy specimen results in a positive diagnosis for amyloidosis, then an accurate typing of the kind of amyloid protein is the next important step.
Other Heart Tests
The echocardiogram (also called “echo”) is an ultrasound of the heart. A doctor can look at the size and shape of the heart, and whether it is relaxing normally in between heartbeats. Amyloid cannot be seen directly, but it does make the heart larger and stiffer than normal.
Other imaging tests for the heart have also shown to be useful. One test is the MRI (magnetic resonance imaging), and, in this instance, is also referred to as CMR (for cardiac magnetic resonance). CMR with a contrast agent called “gadolinium,” given by vein at the time of the scan, is a way to detect amyloid deposits in the heart.
Pyrophosphate scanning, a nuclear medicine test, is also used to evaluate whether an unusual type of abnormality of heart muscle function (“cardiomyopathy”) is present. An intravenous injection of pyrophosphate is made while the patient is at rest, followed about an hour later by a set of images that are taken while the patient lies down under a camera. The images take about 15 minutes. These images are recorded on a computer for analysis, and recent data suggests this scan may be useful in distinguishing different types of amyloid heart disease.
Other imaging tests for the heart have also shown to be useful. One test is the MRI (magnetic resonance imaging), and, in this instance, is also referred to as CMR (for cardiac magnetic resonance). CMR with a contrast agent called “gadolinium,” given by vein at the time of the scan, is a way to detect amyloid deposits in the heart.
Pyrophosphate scanning, a nuclear medicine test, is also used to evaluate whether an unusual type of abnormality of heart muscle function (“cardiomyopathy”) is present. An intravenous injection of pyrophosphate is made while the patient is at rest, followed about an hour later by a set of images that are taken while the patient lies down under a camera. The images take about 15 minutes. These images are recorded on a computer for analysis, and recent data suggests this scan may be useful in distinguishing different types of amyloid heart disease.
How do they diagnose the TYPE of amyloidosis?
If any lab test results in a positive diagnosis for amyloidosis, then identifying the type of amyloid protein is the next crucial step. Treatments can differ and should be tailored to the patient and the exact type of amyloidosis that they have.
Typing can be done by a variety of biochemical and immunologic techniques that include proteomics by mass spectrometry, immunohistochemistry by light or electron microscopy, extraction of proteins with electrophoresis, and others. The accuracy of each form of typing depends upon the technique itself, but also on the ability and experience of the laboratory performing them. It is best to have typing done in a reference laboratory at an experienced center, and the typing should agree with the clinical features of the patient’s disease. Unfortunately, costs of typing are not always covered by insurance, but it is very important to have correct typing before treatment is initiated.
The amyloidosis specialty centers are experts in typing amyloidosis and have the special lab techniques to do this. Some doctors perform the initial testing to determine a positive diagnosis of amyloidosis, however, the biopsy or aspirate sample is often sent to an amyloidosis center to accurately identify the type of amyloidosis.
Typing can be done by a variety of biochemical and immunologic techniques that include proteomics by mass spectrometry, immunohistochemistry by light or electron microscopy, extraction of proteins with electrophoresis, and others. The accuracy of each form of typing depends upon the technique itself, but also on the ability and experience of the laboratory performing them. It is best to have typing done in a reference laboratory at an experienced center, and the typing should agree with the clinical features of the patient’s disease. Unfortunately, costs of typing are not always covered by insurance, but it is very important to have correct typing before treatment is initiated.
The amyloidosis specialty centers are experts in typing amyloidosis and have the special lab techniques to do this. Some doctors perform the initial testing to determine a positive diagnosis of amyloidosis, however, the biopsy or aspirate sample is often sent to an amyloidosis center to accurately identify the type of amyloidosis.
What are the treatment options for AL amyloidosis?
The treatment is tailored to the patient according to their individual heath needs. It is also two-fold and is based upon disease progression and seriousness of the organ, tissue and nerve involvement. The doctor will need to prescribe treatments for the symptoms (supportive treatment) and treatment for the disease itself (source treatment). This is why early diagnosis is crucial, because treatment can have a longer and a more positive outcome when it is started before serious organ damage occurs.
When treating the AL amyloidosis disease itself, the object is to stop (or slow down) the formation of the amyloid light chain protein.
When treating the AL amyloidosis disease itself, the object is to stop (or slow down) the formation of the amyloid light chain protein.
Supportive Treatment
If complications arise from organ damage, then supportive treatment will be necessary. For example, if kidney symptoms increase and renal failure develops, then dialysis may be necessary for the patient. Kidney involvement can be found in approximately 65% of AL amyloidosis patients. If the patient can tolerate the procedure and the kidney damage is severe, a kidney transplant may be an option.
The heart can be involved in up to 74% of AL amyloidosis patients. Blood pressure is often low and cardiac medications can improve heart function. Fluid retention (edema) can be reduced if these symptoms appear. Some patients with heart involvement are at risk for congestive heart failure and experience arrhythmia, which is an irregular heartbeat or abnormal heart rhythm. A heart transplant is also an option for selected patients with severe heart involvement.
Only a few centers around the country are able to consider these types of organ transplants for amyloidosis patients.
It is important to note that if a patient experiences an irregular or unstable heart rhythm and needs immediate attention, the treating doctor must be made aware that the patient has AL amyloidosis. This is because any heart damage in a patient with AL amyloidosis requires a different medical approach than a patient with another type of amyloidosis (or other diseases that may affect the heart). With AL amyloidosis patients, there are certain treatment medications that should be used with caution and gradually administered while being continuously monitored – among them: ACE inhibitors, Beta Blockers, Calcium Channel Blockers and Digoxin.
The heart can be involved in up to 74% of AL amyloidosis patients. Blood pressure is often low and cardiac medications can improve heart function. Fluid retention (edema) can be reduced if these symptoms appear. Some patients with heart involvement are at risk for congestive heart failure and experience arrhythmia, which is an irregular heartbeat or abnormal heart rhythm. A heart transplant is also an option for selected patients with severe heart involvement.
Only a few centers around the country are able to consider these types of organ transplants for amyloidosis patients.
It is important to note that if a patient experiences an irregular or unstable heart rhythm and needs immediate attention, the treating doctor must be made aware that the patient has AL amyloidosis. This is because any heart damage in a patient with AL amyloidosis requires a different medical approach than a patient with another type of amyloidosis (or other diseases that may affect the heart). With AL amyloidosis patients, there are certain treatment medications that should be used with caution and gradually administered while being continuously monitored – among them: ACE inhibitors, Beta Blockers, Calcium Channel Blockers and Digoxin.
Source Treatment
In the United States, a stem cell transplant (SCT) is often the preferred therapy, as it can provide long-term control of the underlying disease. However, only a minority of AL patients are eligible for this. Because such high doses of chemotherapy are used in a SCT, treatment is only given to patients who meet strict criteria. This criteria is based on heart and kidney function and the number of other organs affected by the amyloid protein. After the high dose of chemotherapy is administered, stem cells previously collected from the patient are then put back into the body in order to restore the bone marrow function.
When a stem cell transplant is not possible, a lower dosage of chemotherapy in combination with other drugs is given to destroy the abnormal plasma cells. These drugs are normally given in cycles intravenously (into a vein) or orally (by mouth), over a period of months with rest periods in between to allow the cells to recover.
The types of drugs that are used are:
When a stem cell transplant is not possible, a lower dosage of chemotherapy in combination with other drugs is given to destroy the abnormal plasma cells. These drugs are normally given in cycles intravenously (into a vein) or orally (by mouth), over a period of months with rest periods in between to allow the cells to recover.
The types of drugs that are used are:
- Corticosteroids – Dexamethasone and Prednisone Alkylating agents – MelphalanCyclophosphamide (Cytoxan™)
- Bendamustine (Treanda™) Immunomodulatory drugs – Thalidomide (Thalidomid™), Lenalidomide (Revlimid™)
- Pomalidomide (Pomalyst™) Proteasome inhibitors – Bortezomib (Velcade™), MLN9708 (Ixazomib™)
- Carfilzomib (Kyprolis™)
Examples of some chemotherapy combinations include:
- Melphalan & Dexamethasone
- Cyclophosphamide (Cytoxan™) & Bortezomib (Velcade™) & Dexamethasone
- Cyclophosphamide (Cytoxan™) & Thalidomide (Thalidomid™) & Dexamethasone
- Cyclophosphamide (Cytoxan™) & Lenalidomide (Revlimid™) & Dexamethasone
- Bortezomib (Velcade™) & Dexamethasone Melphalan & Prednisone & Lenalidomide (Revlimid™)
Note that some of these drugs are still considered investigational for AL amyloidosis, and should not be used outside of a sponsored clinical trial. Also, most of them are given with a steroid such as dexamethasone, which seems to increase their effectiveness.
Is there a special diet that I can follow?
Eating a well-balanced, heart-healthy and nutritious diet is always recommended. Although amyloid is an abnormal protein, the amount of protein in the diet does not affect the onset of the disease. A diet low in protein and/or sodium may be necessary when the kidneys or heart are involved. Consult with your physician on any dietary changes, and report any vitamins or other supplements that you take. You are a part of the team of people who must keep in communication with each other about your health.
What kind of doctor should be consulted?
It is strongly recommended that you consult with a specialist in the field of amyloidosis. The Amyloidosis Foundation provides a list of amyloidosis treatment centers under “Patient Resources” on this website. Once your diagnosis is confirmed, then a treatment plan can be laid out for your individual case. Depending on your symptoms, you will be seeing a local hematologist (blood), oncologist (cancer), neurologist (nerves), cardiologist (heart), nephrologist (kidney), gastroenterologist (GI tract), internist and/or general physician. These doctors should coordinate your care with the amyloidosis specialist to develop the best treatment program.