AA amyloidosis occurs as a reaction to another illness, such as a chronic inflammatory disease or a chronic infection. Infections and inflammation cause the liver to produce a protein called SAA (serum amyloid A protein) at high levels. This is a normal reaction.
When inflammation goes on for a very long period of time, a small portion of the SAA protein, called AA protein, will separate from SAA and deposit in tissues as AA amyloid. Normally after an inflammatory reaction, the whole SAA protein is broken down to its amino acid components and recycled, as happens with all proteins. It is not known why in some individuals a partial breakdown of SAA to AA occurs.
These individuals usually have severe and chronic inflammatory conditions lasting several years. They develop AA amyloid deposits in all tissues, but the most common organ damage occurs in the kidneys. Some patients experience complications with their liver, spleen, thyroid, digestive tract, or heart. Any chronic inflammation that elevates the SAA protein for a long time has the potential to lead to AA amyloidosis.
Inflammatory diseases that are more commonly known to lead to AA amyloidosis are in the following categories. Rheumatologic diseases include rheumatoid arthritis, juvenile arthritis, ankylosing spondylitis, and psoriatic arthritis. Gastrointestinal inflammatory diseases, including Crohn’s disease and ulcerative colitis.
Chronic Infections, such as tuberculosis, bronchiectasis, osteomyelitis, or infections associated with cystic fibrosis, AIDS, and needle-using drug addiction that cause skin infections. Hematologic malignancies, including Hodgkin’s disease, renal cell carcinoma, and Castleman’s disease. Hereditary disorders that cause disruption of inflammatory genes, such as Familial Mediterranean Fever (FMF), TRAPS (tumour necrosis factor receptor-associated periodic syndrome), and others.
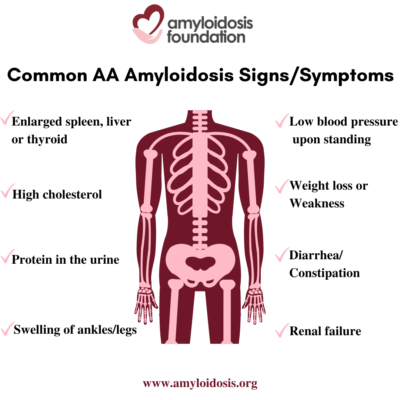
Symptoms
The term “nephrotic syndrome” refers to a group of symptoms that signal kidney problems. The first symptom of AA amyloidosis is usually protein in the urine. Often proteinuria (protein in the urine) becomes massive, and nephrotic syndrome develops. This means that patients are losing so much protein in their urine that they develop low levels of protein (albumin) in their blood, leading to swelling of the ankles and legs (edema). A high cholesterol level is also part of this syndrome. As a result, patients may develop renal failure and the need for dialysis.
AA amyloidosis involves other organs in addition to the kidneys. An enlarged spleen, enlarged liver, and enlarged thyroid are not uncommon. Autonomic neuropathy is frequent with symptoms of orthostatic hypotension (low blood pressure on standing), gastrointestinal atony (slowing of stomach emptying) and diarrhea or constipation. AA amyloid deposits in the heart causing congestive heart failure and arrhythmias (irregular heart beat) may develop later in the course of the disease.
Symptoms in a patient with AA amyloidosis can be misunderstood as symptoms that relate to their chronic infection or inflammation. At first, a patient may have symptoms such as weight loss, weakness, and swelling (edema). Since a patient’s primary disorder may also cause these problems, a doctor may assume that these are a result of their primary condition. This can delay further testing which would lead to a diagnosis of the secondary disease, AA amyloidosis.
In a reverse situation, AA amyloidosis may be found first, before another disease or condition is identified. For example, AA amyloidosis may be diagnosed as a result of nephrotic syndrome and then may lead to the investigation for an undiagnosed, underlying condition. So, although the AA amyloidosis was triggered by a primary disorder, it does not always mean that the primary disorder was previously discovered and diagnosed.
Diagnosis
If a patient has previously been diagnosed with a chronic inflammatory disease or chronic infection and they develop high levels of protein in the urine or other associated AA symptoms, then the physician should test for AA amyloid deposition. When renal damage occurs, it can be clinically shown as proteinuria (protein found in the urine), nephrotic syndrome, or impairment of renal (kidney) function.
A test involving a 24-hour urine collection can be performed to look at the level of protein in the patient’s urine sample. Protein in the urine is an indication of kidney complications. If amyloidosis is suspected through this and other test results and associated symptoms, in most cases a biopsy of renal (kidney) tissue is recommended to get an accurate diagnosis.
This renal biopsy tissue is sent to a lab for Congo-red staining. The lab will stain the biopsy and, if it turns an apple green color under a ‘polarizing’ microscope, then amyloidosis is confirmed. Once this initial diagnosis has been determined, it is very important to find out the exact protein type in a positive tissue biopsy so that appropriate treatment can be recommended.
In order to identify the amyloid type, the most common diagnostic test is staining the tissue sample with antibodies that are specific for the major amyloid protein diseases, such as “anti-AA serum,” AL light chains, and anti-TTR. If it is the anti-AA serum result that is positive in this lab test, then AA amyloidosis is diagnosed. It is important to rule out other types because other amyloid diseases may involve the kidneys and those patients may also present with a high level of protein in their urine. However, another type of amyloid disease that is known for kidney involvement, such as AL amyloidosis, would require a different treatment regimen.
Once AA amyloidosis is confirmed it is important to look for the primary underlying inflammatory condition, if not already known. Then, the next step is to determine the extent of amyloid involvement in all organs and develop a plan for treatment. This is done by a careful physical examination, and laboratory studies of kidney and heart function.
Treatment
Each amyloidosis type has a specific treatment. Early detection and timely treatment is a key factor. The type of treatment is based upon disease progression and seriousness of the patient’s organ, tissue and/or nerve involvement.
AA amyloidosis treatment plans include:
- Underlying disease treatment – continual management of the primary disease;
- Supportive treatment – treating patient symptoms and organ damage; and,
- Amyloid source treatment, when available – slowing down, or stopping, the overproduction of amyloid at the source of this secondary disease.
Underlying disease treatment
In AA amyloidosis, the most important therapy is to treat the underlying infection or inflammation in order to reduce the level of SAA protein, the precursor for the AA amyloid deposits. These treatments will vary depending on the underlying condition. The effectiveness of this treatment can be monitored by blood tests that measure inflammation in the blood, such as sedimentation rate and C-reactive protein levels. Some treatments that exist for inflammatory diseases could include surgery on the infection or tumor; drug therapies for rheumatoid arthritis; antibiotics for chronic infection; and the use of colchicine for FMF (Familial Mediterranean Fever), among others.
With effective treatment of the underlying inflammation amyloid deposits have been known to reduce and nephrotic syndrome can improve. However, if the kidney function has become significantly impaired, it rarely recovers.
Supportive treatment
Supportive treatment is very important for patients with AA amyloidosis. A team of specialists, including a nephrologist, cardiologist, and neurologist in addition to the primary physician is helpful to manage the disease development in the various organ systems.
Kidney damage is often a major health issue associated with AA amyloidosis. Regular blood and urine tests are recommended to monitor the patient’s renal (kidney) function. Managing proteinuria (protein in the urine) and nephrotic syndrome and keeping renal function from declining can delay the need for dialysis and improve the quality of life. Under the close supervision of the doctor, the supportive treatment that is often helpful for nephrotic syndrome includes an increase of protein in the diet, monitoring salt intake, and use of support hose. Other things that are very important when the kidney has amyloid deposits are maintaining normal blood pressure and avoiding dehydration.
It is important to note that the patient should not take any medication (prescription or over the counter) unless it has been approved by the nephrologist. When the kidney already has some damage, some medications can worsen this kidney damage. It can happen even when these same medications would be acceptable for normal kidneys. Therefore, it is very important that all patients with AA amyloidosis and renal involvement have a nephrologist on their medical team.
Dialysis is an option for patients with AA amyloidosis that are in renal failure, especially when other organ function is in good condition. A kidney transplant is an option in some cases, particularly if the associated inflammatory disease or chronic infection has been treated successfully.
Supportive treatment for autonomic neuropathy includes maintaining blood pressure when standing by using support hose, along with a slight increase of salt in the diet or medications that raise blood pressure.
Symptoms of gastric distress may be harder to manage, but frequent small meals and a diet lower in fat are often helpful. Medications can be used for diarrhea. An adjustment of the patient’s meal timing can also ease the onset of diarrhea.
Cardiac involvement needs to be watched closely by a cardiologist to manage treatment for congestive heart failure or arrhythmia.
FAQ
FAQ
Why is it called AA amyloidosis?
In the past, AA amyloidosis was referred to as “Secondary” or “Inflammatory” amyloidosis. These are no longer accepted names for this form of amyloidosis, which is usually caused by a complication of chronic inflammation or chronic infection. If a patient has chronic inflammation or chronic infection due to a number of possible conditions or diseases, this inflammation can often trigger an increased production of the SAA (Serum Amyloid A) protein in the body. When inflammation goes on for a very long period of time, a small portion of the SAA protein, called AA protein, will separate from SAA. This AA amyloid protein can then misfold, causing amyloid fibrils that clog and interfere with tissue and organ function.
Since systemic amyloidoses are referred to with a capital A (for amyloid) followed by an abbreviation for the fibril protein, the second “A” in AA amyloidosis stands for the fragment AA protein of Serum Amyloid A (SAA).
What is SAA protein?
SAA stands for Serum Amyloid A protein. The progression and severity of AA amyloidosis relates to the production and quantity of the SAA protein that is produced in that patient. One medical dictionary definition defines it as: “A high-molecular-weight protein synthesized in the liver; it is an acute phase protein and circulates in association with HDL lipoprotein. It is the precursor to AA amyloid and accumulates in inflammation.”
In simpler terms, even though SAA is mainly produced in the liver, it circulates in the blood and is composed of different forms of proteins. These different forms of proteins assume several roles in the body, including carrying cholesterol to some organs and signaling germ-fighting cells (immune cells) to travel to areas of infection or inflammation. Therefore, an increase in SAA protein in the body is often a response to a disease or condition.
What is chronic inflammation?
It is the body’s chronic (long term and continuing) response to disease, infection or injury. If a disease or an injury occurs, the body’s immune system fights it and, once the response and repair is successful, the inflammation is under control and no tissues or cells can continue to be damaged. However, if the inflammation does not stop and it continues, then the cells may change in that area and they may loop – causing damage, healing, and damage again. Over a long period of time, this may result in a breakdown of the tissues or organs and can cause progressive damage.
Medical conditions that end in “itis” are linked with inflammation. For example, when considering chronic inflammation, one might think of arthritis, which can result in inflammation in a person’s joints, causing swelling and pain. Another example is osteomyelitis, which is an infection and inflammation of the bone or bone marrow.
Markers can be detected in blood tests and can help with monitoring chronic inflammation. “C-reactive protein” is one of the key markers.
What is nephrotic syndrome?
Nephrotic syndrome is a kidney disorder. It represents a group of symptoms and is a signal that your kidneys are not working properly. Signs of nephrotic syndrome include protein in the urine (proteinuria), low blood protein, and high cholesterol. In addition, patients may experience these symptoms:
- fatigue;
- high blood pressure;
- edema (swelling), especially in the ankles, feet, and face;
- foamy urine (due to excess protein);
- unexplained weight gain (due to fluid retention);
- loss of appetite; and,
- malnutrition (due to loss of too much blood protein).
Proper medical treatment along with a change in the patient’s diet may help to control the symptoms. If nephrotic syndrome worsens and the kidneys sustain more damage, then a patient may develop renal failure and the need for dialysis.
What tests might help with determining organ function?
Doctors may diagnose nephrotic syndrome using urine tests, blood tests, and/or ultrasound to look at the kidneys. Sonography is useful to establish and monitor the size of the kidneys. However, it’s important to note that everybody is different and patients that have AA amyloidosis with renal dysfunction may have “normal” kidneys of various sizes – their kidneys may originally be small, large or anywhere in between. The doctor will benefit from having a comparison “baseline” of that patient’s kidney size in order to monitor renal changes through sonography.
A small percentage of AA amyloidosis patients have symptoms that indicate cardiac (heart) involvement, and blood tests for heart biomarkers can aid in determining if a patient has signs of heart tissue strain or damage in their blood. The results of these tests can be used as “markers” (also called “biomarkers”) to first determine the extent of any damage, and then can be used regularly to monitor any future problems. It is possible that these biomarker blood tests may be affected by changes in kidney function and other causes, so they should be interpreted in combination with other tests of cardiac function, such as an echocardiogram or CMRI (cardiac magnetic resonance imaging).
What is autonomic neuropathy and what are the symptoms?
Autonomic neuropathy (AN) is a condition that results from damage to nerves that assist in organ and organ system functioning. Autonomic nerves control the functions of our internal organs such as the heart, stomach and intestines, as well as the glands. When your autonomic nerves are damaged, your blood pressure, heart rate, perspiration patterns, and bowel movements may be affected. In addition, you may have problems with dizziness, emptying your bladder, and/or experience gastrointestinal symptoms of diarrhea, weight loss, and poor digestion.
At what age can you get AA amyloidosis?
AA amyloidosis can occur at any age and is the only amyloidosis to occur in children. Since AA amyloidosis is a reactionary disease to other diseases or conditions, the age of onset for AA amyloidosis depends on when the patient develops a chronic inflammatory disease or chronic infection. It is also related to the severity and duration of that disorder.
For example, juvenile rheumatoid arthritis (JRA) can occur at age 16 years or younger. It is possible that AA amyloidosis may appear in children or teenagers, although AA amyloidosis symptoms don’t often occur until long-term complications from JRA are underway. With adult Rheumatoid Arthritis (RA), AA amyloidosis may develop in late middle age or later. If Familial Mediterranean Fever (FMF) is not treated effectively, it is more common for AA amyloidosis to develop sooner in the course of the disease, with renal complications. So, there is no standard age range for AA amyloidosis.
Is it a form of cancer?
No. At this time, none of the types of amyloidosis diseases are considered to be cancer.
How common is AA amyloidosis?
AA amyloidosis is considered a rare disease. In all areas of the world, the frequency of AA amyloidosis depends on the presence and severity of the associated and underlying disorder. These underlying disorders are, more and more, diagnosed earlier and treated aggressively. The World Health Organization (WHO) offers multidrug therapy free of charge and as result leprosy is more controlled, except in a few countries. Effective antibiotics have decreased the incidence of chronic infections, including tuberculosis. Since 1974, colchicine has been used to treat Familial Mediterranean Fever and, as a result, fewer patients with FMF develop AA amyloidosis. New immunomodulary drugs are used for rheumatic diseases that have decreased inflammation dramatically, and thus secondarily decreased AA amyloidosis.
AA amyloidosis is not as common in the United States and some European countries when compared with many of the underdeveloped countries. All of the reasons for this are not completely understood. Some Third World countries may not have the same medical or lab facilities or availability of drug therapies for the associated disorders. Geographic locations may play a part when considering the genetic backgrounds of the population, the environmental and economic factors, along with the living conditions of the people in any given country.
Statistics still vary regarding the frequency of AA amyloidosis and research continues.
What are the diseases and conditions that can trigger AA amyloidosis?
Do not presume that if you have any of these conditions that AA Amyloidosis will develop. Patients can live with a chronic inflammation or chronic infection and not be diagnosed with AA amyloidosis.
Currently, the rheumatic diseases such as Rheumatoid Arthritis (RA), Ankylosing Spondylitis (AS), Psoriatic Arthritis, and Juvenile Arthritis represent a majority of the causes of AA amyloidosis. Other more commonly known conditions that can trigger AA Amyloidosis are: Inflammatory Bowel Disease (IBD), Crohn’s Disease, Familial Mediterranean Fever (FMF), Hodgkin’s Lymphoma, Tuberculosis, Renal Cell Carcinoma, and HIV/AIDS.
The long list below includes what some of the medical community links to the disorders associated with AA amyloidosis. Some of these underlying disorders are very rare and we have found that each list varies among specialists.
Inflammatory Arthritides
- Rheumatoid Arthritis
- Ankylosing Spondylitis
- Adult Still’s Disease
- Juvenile Idiopathic Arthritis
- Psoriatic Arthritis
- Gout
Inflammatory Bowel Diseases (IBD)
- Crohn’s Disease
- Ulcerative Colitis
Systemic Vasculitides
- Behcet’s Disease
- Polyarteritis Nodosa
- Giant Cell Arteritis
- Takayasu’s Arteritis
- Polymyalgia Rheumatica
Hereditary Autoinflammatory Diseases
- Familial Mediterranean Fever (FMF)
- Tumor Necrosis Factor Receptor Associated Periodic Syndrome (TRAPS)
- Muckle–Wells Syndrome
- Neonatal-onset Multisystem Inflammatory Disease (NOMID)/Chronic Infantile Neurological, Cutaneous and Articular (CINCA) Syndrome
- Hyper-IgD Syndrome
Neoplasms
- Castleman’s Disease
- Hodgkin’s Lymphoma
- Waldenström’s Macroglobulinemia
- Hairy Cell Leukemia
- Hepatocellular Adenoma
- Renal Cell Carcinoma
- Adenocarcinoma of the lung
- Adenocarcinoma of the gut
- Mesothelioma
- Schnitzler Syndrome
Chronic infections
- Osteomyelitis
- Tuberculosis
- Pyelonephritis
- Leprosy
- Whipple’s Disease
Conditions predisposing to chronic infections
- Bronchiectasis
- Chronic Cutaneous Ulcers
- Cystic Fibrosis
- Epidermolysis Bullosa
- Injection drug users (long-term intravenous drug use, or IVDU)
- Jejuno-ileal Bypass
- Paraplegia
Hereditary and acquired immunodeficiencies
- Common Variable Immunodeficiency
- Hypogammaglobulinemia
- X-linked Agammaglobulinemia
- Cyclic Neutropenia
- HIV infection/Acquired Immunodeficiency Syndrome (AIDS)
Other
- Obesity
- Sarcoidosis
- Synovitis Acne Pustulosis Hyperostosis
- Osteitis (SAPHO) Syndrome
Is there a special diet that I can follow?
Eating a well-balanced, heart-healthy and nutritious diet is always recommended. Although amyloid is an abnormal protein, the amount of protein in the diet does not affect the onset of the disease. However, a diet low in protein and/or sodium may be necessary when the kidneys are involved, and fluid intake should be steady and not excessive. It can be helpful to meet with a medically trained dietician for individual and personal advice to address your exact symptoms and needs. Consult with your physician on any dietary changes, and report any vitamins or other supplements that you take. You are a part of the team of people who must keep in communication with each other about your health.
What kind of doctor should be consulted?
It is strongly recommended that you consult with a specialist in the field of amyloidosis. The Amyloidosis Foundation provides a list of amyloidosis treatment centers under “Patient Resources” on this website. Once your diagnosis is confirmed, then a treatment plan can be laid out for your individual case. Depending on your symptoms, you will be seeing a local hematologist (blood), oncologist (cancer), neurologist (nerves), cardiologist (heart), nephrologist (kidney), gastroenterologist (GI tract), internist and/or general physician. These doctors should coordinate your care with the amyloidosis specialist to develop the best treatment program.